
Sommaire
Introduction
Vocabulaire
Loi des gaz parfaits
Isochore – Isobare – Isotherme
Calcul de Q
Calcul de W
Premier principe de la thermodynamique
L’enthalpie
La capacité calorifique
Transformations adiabatiques : formules de Laplace
Changement d’état et diagramme de Clapeyron
Annexe : forces de pression
Exercices
La thermodynamique est un domaine assez complexe que nous allons donc découper en plusieurs parties. Le chapitre ci-dessous détaille tout le vocabulaire spécifique à la thermodynamique ainsi que le premier principe de la thermodynamique.
Avec tout cela tu pourras déjà faire pas mal d’exercices ! 
Volontairement, il n’y a pas beaucoup d’exemples d’application dans ce chapitre afin de ne pas surcharger et de se concentrer sur les formules et les propriétés, mais un autre chapitre sera entièrement consacré à des applications simples du cours, qui peuvent être considérées comme des exemples.
La thermodynamique décrit l’évolution de systèmes qui subissent des échanges d’énergies avec l’extérieur.
Il y a donc, comme en mécanique par exemple, la notion de système. Tout ce qui est en dehors du système est considéré comme le milieu extérieur. Il faudra donc faire attention à choisir correctement ce système afin de répondre aux questions.
Ce système possède des grandeurs dites extensives, et d’autres intensives.
Pour faire la différence entre les 2, prenons un exemple très simple.
Imaginons que l’on ait 2 pièces l’une à côté de l’autre, séparées par une cloison amovible.
Chacune à un certain volume, une certaine température, pression etc…
Si on enlève la cloison, le volume total sera la somme des 2 volumes, ce qui ne sera pas le cas pour la température !
Le volume est donc une variable dite extensive, la température est une variable dite intensive.
Exemples de variables extensives : la masse, la quantité de matière…
Exemples de variables intensives : la pression, la concentration…
Le système étudié sera par ailleurs défini par certaines variables appelées variables d’état (P, T, V etc…), nous en reparlerons plus loin.
Un système est dit en équilibre quand ses variables sont constantes au cours du temps.
On parle d’équilibre thermodynamique quand le système est en équilibre thermique, mécanique et chimique. Dans ce chapitre, on ne se préoccupera pas de l’équilibre chimique.
Nous verrons des exemples dans le chapitre sur les applications du 1er principe.
Sous l’effet d’échanges d’énergie entre le système et le milieu extérieur, cet équilibre initial va être rompu et ces variables vont évoluer vers un nouvel état d’équilibre : le but sera de déterminer cette évolution grâce aux différentes équations et principes détaillés ci-dessous.
Ces échanges énergétiques entre le système et le milieu extérieur seront modélisés de 2 manières différentes : du travail, noté W, et du transfert thermique noté Q.
Q est aussi appelée la chaleur (attention, la chaleur ne correspond pas à la température contrairement au langage courant).
W sera généralement du travail mécanique, ou électrique mais c’est plus rare.
W et Q étant des énergies, elles seront bien évidemment en Joules.
On peut schématiser cela de la manière suivante :
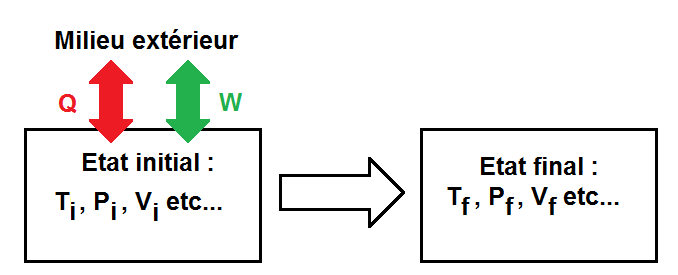
Ti est la température initiale, Pi la pression initiale, Vi le volume initial etc…
Le système peut bien sûr donner ou recevoir de l’énergie du milieu extérieur. Pour faire la différence entre les deux, W et Q seront positifs ou négatifs selon que le système donne ou reçoit de l’énergie.
Mais dans quel cas Q et W sont-ils positifs ou négatifs ?
Pour s’en souvenir c’est extrêmement simple, le principe est le même que pour l’argent !
Si tu reçois 1 euro par exemple, tu auras +1 euro sur ton compte, si au contraire tu donnes 1 euros tu auras -1 euro sur ton compte.
Tu l’auras compris :
—
Si le système reçoit de l’énergie, W > 0, Q > 0
Si le système donne de l’énergie, W < 0, Q < 0
—
Evidemment il peut y avoir différents échanges d’énergie pour une même transformation, on peut donc par exemple avoir W1 > 0, W2 < 0 et Q1 < 0 :
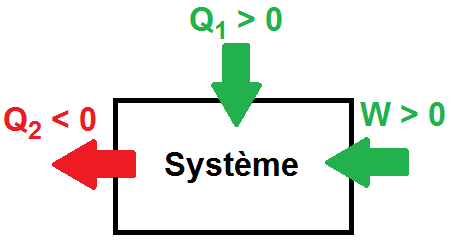
Si on reprend le principe de l’argent, si tu gagnes 1 euro tu auras +1 sur ton compte, mais la personne qui t’a donné 1 euro aura quant à elle -1 sur son compte !
Ainsi, un même échange peut être positif ou négatif selon le point de vue duquel on se place, d’où l’importance de bien choisir le système !
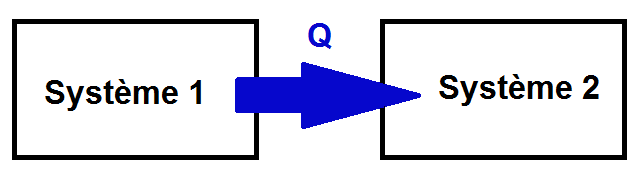
Dans l’exemple ci-dessus, Q < 0 pour le système 1 mais Q > 0 pour le système 2 : tout dépend du système !
Mais comment savoir dans quel sens se fait le transfert thermique ?
Soit cela est donné dans l’énoncé, soit on applique un principe très simple : le transfert thermique se fait toujours du corps le plus chaud vers le corps le plus froid. C’est ce que l’on appelle l’énoncé de Clausius.
Ainsi, s’il fait plus chaud en dehors de la maison qu’à l’intérieur par exemple, le transfert thermique se fera de l’extérieur vers l’intérieur.
Certaines transformations seront dites adiabatiques, cela signifie que Q = 0 : il n’y a pas d’échange thermique, le système est isolé thermiquement (mais il peut y avoir du travail W).
Attention, cela ne signifie pas que la température est constante !!
De plus, une paroi est dite diathermane si elle permet les échanges thermiques. Une paroi athermane au contraire empêche tout échange d’énergie thermique.
Ainsi, pour une boîte aux parois athermanes contenant un gaz, si on prend comme système le gaz, Q = 0 (car pas d’échange thermique), et donc toute transformation sera adiabatique.
En résumé :
—
Une transformation est dite adiabatique si Q = 0
Une paroi est dite diathermane si elle permet des échanges thermiques avec l’extérieur
Une paroi est dite athermane si elle ne permet pas d’échanges thermiques avec l’extérieur
—
Remarque : tu verras parfois le terme diathermique à place de diathermane, c’est la même chose !
S’il y a des échanges d’énergie, cela signifie que le système a une certaine énergie, que l’on appelle l’énergie interne, notée U.
Cette énergie est évidemment en Joules.
Nous y reviendrons en détail plus tard.
Bien que ce soit toutes des énergies, il existe une grosse différence entre U d’une part, et Q et W d’autre part.
En effet, U est ce que l’on appelle une fonction d’état, c’est-à-dire une fonction dont la variation ne dépend pas du chemin suivi.
La variation d’une telle variable est notée avec Δ (prononcer delta).
Ainsi la variation d’énergie interne au cours d’une transformation sera notée ΔU.
Il en est de même pour l’enthalpie H et l’entropie S dont nous parlerons ultérieurement : ΔH et ΔS.
En revanche, Q et W dépendent du chemin suivi par la transformation, ΔQ et ΔW n’ont aucune signification physique, donc à ne surtout pas écrire sur une copie !!!
—
Remarque : le principe de fonction d’état s’apparente un peu à celui de force conservative en mécanique. En effet, en mécanique, une force est conservative si son travail est indépendant du chemin suivi.
Ici, une grandeur est une fonction d’état si sa variation est indépendante du chemin suivi.
—
De plus, il est important de faire la différence pour le calcul avec des grandeurs infinitésimales.
En effet, la variation des grandeurs physiques se calculera parfois (et même souvent) en intégrant une variation infinitésimale de la grandeur en question.
Exemples :
\(\textstyle \Delta U = \int dU \)
\(\textstyle \Delta H = \int dH \)
\(\textstyle Q = \int dQ \)
\(\textstyle W = \int dW \)
Comme tu le vois, quand on intègre une variable, on trouve la variation de cette grandeur, mais qui n’est pas notée avec Δ pour Q et W car ce ne sont pas des fonctions d’état.
Enfin, on a dit précédemment que l’on allait étudier un système évoluant d’un état initial à un état final.
Mais peut-on, à partir de l’état final, revenir à l’état initial ?
Si tel est le cas, on dit que la transformation est réversible, sinon elle est dite irréversible.
Mais pour que la transformation soit réversible, il faut qu’elle soit infiniment lente, et qu’elle passe par une infinité d’états intermédiaires considérés comme des états d’équilibre.
Une transformation réversible n’existe pas en réalité, mais on peut modéliser certaines transformations ainsi, il s’agit donc d’un cas idéal.
En résumé :
—
Transformation réversible : transformation infiniment lente, succession d’états d’équilibre : on parle aussi de transformation quasi-statique.
Transformation irréversible : les autres transformations.
—
La loi des gaz parfaits est une loi très simple utilisée dans de nombreux exercices.
Elle ne s’applique, comme son nom l’indique, qu’aux gaz parfaits ou supposés parfaits : il est presque toujours dit dans l’énoncé que tel gaz est considéré comme parfait (parfois il faut en faire l’hypothèse mais c’est très rare).
Cette loi est la suivante :
\(\displaystyle PV = nRT \)
Dans cette formule :
P : pression en Pascal (Pa). Attention, la pression est souvent donnée en bars : 1 bar = 105 Pa
V : volume en m3. Souvent à convertir car donnée en L ou en mL. Pour savoir comment convertir les volumes, clique ici
n : nombre de moles (en mol)
R : constante des gaz parfaits : 8,314 J.K-1.mol-1
T : température en Kelvins (K). Attention la température est souvent donnée en degrés celsius : T(K) = T(°C) + 273,15
—
ATTENTION surtout aux unités dans cette formule : P, V et T sont souvent à convertir.
Donc ne pas oublier que P est en Pa, V en m3 et T en K.
—
Si un gaz n’est pas parfait, on utilise parfois l’équation de Van der Walls :
\(\textstyle (P + \frac{an^2}{V^2})(V – nb) = nRT \)
a et b sont des coefficients : s’ils sont nuls, on retrouve la loi des gaz parfaits.
Il existe certains exercices utilisant cette formule mais c’est assez rare donc nous ne rentrerons pas dans les détails. Cette formule n’est pas à savoir.
—
Remarque : la pression est parfois exprimée en mmHg : millimètre de mercure.
C’est une autre unité de la pression, qu’il faudra évidemment convertir en Pa pour les formules.
La conversion est : 1 mmHg = 133,322 Pa. Ce n’est pas à savoir mais on ne sait jamais…
—
Certaines transformations seront isothermes, isobares ou isochores.
C’est généralement bon signe car cela va simplifier les calculs !
En effet, isotherme signifie que la température du système est constante.
Isobare signifie que la pression du système est constante.
Isochore signifie que le volume du système est constant.
Ainsi, en notant f les variables de l’état final et i celles de l’état initial :
Pour une transformation isotherme, Tf = Ti.
Pour une transformation isobare, Pf = Pi.
Pour une transformation isochore, Vf = Vi.
Il est alors intéressant de tracer ces transformations dans ce que l’on appelle un diagramme de Clapeyron, aussi appelé diagramme (P , V).
Attention, on dit diagramme (P , V) mais c’est le V en abscisse et le P en ordonnée :

Dans un tel diagramme, une transformation isobare est donc représenté par une droite horizontale, et une isochore pour une droite verticale.
Mais une isotherme ?? 
Si on considère la transformation d’un gaz parfait, on a :
\(\textstyle P = \frac{nRT}{V} \)
n, R et T étant constants (T est constant car isotherme), V étant x dans le graphe et P étant y (abscisse et ordonnée), on a une équation du type :
\(\textstyle y = \frac{K}{x} \)
Une isotherme est donc représentée par une partie de fonction inverse, c’est-à-dire une hyperbole :
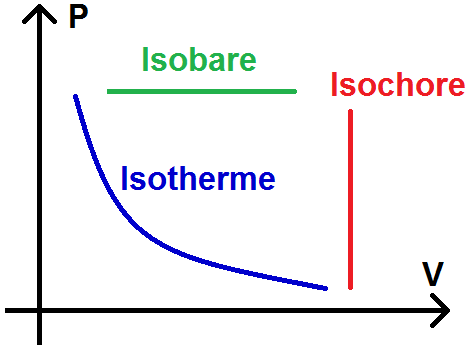
Attention cependant à ne pas confondre avec monotherme et monobare !!
En effet, une transformation monotherme signifie que la température extérieure est constante.
Monobare signifie que la pression extérieure est constante.
Alors que pour isotherme, isobare et isochore, c’est la grandeur du système (et non celle du milieu extérieure) qui est constante.
Remarque : monochore ne veut pas dire grand chose, car alors le volume extérieur serait constant, ce qui n’a pas de sens…
En résumé :
—
Iso-… signifie que la grandeur du système est constante
Mono-… signifie que la grandeur extérieure est constante
—
Q va se calculer essentiellement de 2 manières, correspondant à 2 transformations distinctes.
Tout d’abord, quand il y a un changement de température.
On a la formule suivante :
\(\displaystyle dQ = mcdT \)
Dans cette formule :
dQ est la variation infinitésimale de chaleur (en J)
m la masse du système (en kg)
dT la variation infinitésimale de température (en K)
c la capacité calorifique massique (en J.kg-1.K-1)
Cette capacité calorifique peut aussi être molaire (notée cm) ou bien ni massique ni molaire et notée dans ce cas C.
On a alors les formules :
\(\textstyle dQ = nc_mdT \)
avec n la quantité de matière
\(\textstyle dQ = CdT \)
Mais dans la plupart des exercices on a la massique, d’où l’intérêt de retenir plutôt la 1ère formule.
Remarque : la capacité calorifique massique est notée c en minuscule, la non massique avec C majuscule pour ne pas confondre.
Attention aussi, le m de cm signifie molaire et non massique…
Mais il peut arriver que les notations soient différentes dans certains énoncés, donc attention !
Pour calculer Q, il n’y a plus qu’à intégrer :
\(\textstyle Q = \int dQ \)
\(\textstyle Q = \int mc dT \)
Si c ne dépend pas de T :
\(\textstyle Q = mc \int dT \)
\(\textstyle Q = mc \Delta T \)
Si c dépend de T, il faudra évidemment le remplacer par son expression et calculer l’intégrale.
A noter que c peut être à volume constant ou à pression constante (notés cV et cP), nous y reviendrons ultérieurement.
L’autre manière de calculer Q est d’utiliser la chaleur latente de changement d’état.
Cela est possible quand le fluide change d’état. Les 6 transformations possibles sont les suivantes :
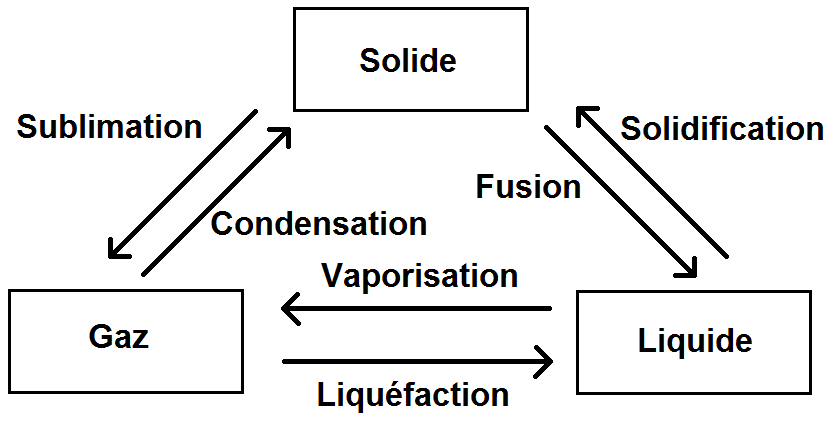
Il existe, pour chaque corps, 3 valeurs correspondant au transfert thermique de changement d’état, notées L avec en indice la première lettre de la transformation :
Ls : chaleur latente massique de sublimation (en J.kg-1)
Lv : chaleur latente massique de vaporisation (en J.kg-1)
Lf : chaleur latente massique de fusion (en J.kg-1)
Pour calculer le transfert thermique, il suffit de multiplier cette chaleur par la masse qui subit le changement d’état, on a donc :
\(\displaystyle Q = mL \)
Suivant la transformation subie, on choisira le bon L.
Mais comment faire si la transformation n’est ni une sublimation, ni une vaporisation, ni une fusion ?
Le principe est très simple :
—
La chaleur massique associée à un changement d’état est l’opposée de celle associée au changement d’état inverse.
—
Autrement dit :
\(\displaystyle L_{condensation} = -L_{sublimation} \)
\(\displaystyle L_{liquefaction} = -L_{vaporisation} \)
\(\displaystyle L_{solidification} = -L_{fusion} \)
C’est la raison pour laquelle il n’y a que Ls, Lv et Lf, les 3 autres se déduisent de ces 3 là.
A noter que Ls, Lv et Lf sont positifs (ils correspondent à une augmentation de température), tandis que les autres sont négatifs (diminution de température).
Après avoir vu comment calculer la chaleur, voyons comment calculer W.
Le travail se calcule grâce à la relation :
\(\displaystyle dW = -PdV \)
dans cette formule :
dW est la variation infinitésimale de travail (en J)
P est la pression (en Pa)
dV est la variation infinitésimale de volume (en m3)
Le signe – vient du fait que si le volume augmente (dV > 0), cela signifie que le système a fourni un travail pour augmenter sa taille, donc dW < 0. La pression P étant positive, le signe - s'impose. Pour calculer W, il suffit d'intégrer :
\(\textstyle W = \int dW \)
\(\textstyle W = \int -PdV \)
Mais contrairement à Q où le calcul est souvent simple, ici il faudra développer un peu plus les calculs car P n’est pas indépendant de V (sauf dans certains cas que nous allons voir).
Voyons les 3 cas les plus fréquemment rencontrés et évoqués précédemment : les transformations isochores, isobares et isothermes.
– transformations isochores : V = constante
\(\textstyle W = \int -PdV \)
\(\textstyle W = 0 \)
dV = 0 car V est constant
– transformations isobares : P = constante
\(\textstyle W = \int -PdV \)
\(\textstyle W = -P\int dV \)
car P est constant
\(\textstyle W = -P\int\limits_{V_1}^{V_2} dV \)
\(\textstyle W = -P(V_2 – V_1) \)
\(\textstyle W = -P\Delta V \)
—
Remarque : les bornes de l’intégrale sont V1 et V2 car la variable est V. Si on avait eu dP, on aurait intégré de P1 à P2, et si on avait eu dT, on aurait intégré de T1 à T2.
—
– transformations isothermes : T = constante
\(\textstyle W = \int -PdV \)
On va considérer que l’on a un gaz parfait, donc PV = nRT, d’où :
\(\textstyle P = \frac{nRT}{V} \)
En remplaçant :
\(\textstyle W = \int -\frac{nRT}{V} dV \)
n, R et T étant constants (car transformation isotherme) :
\(\textstyle W = -nRT \int \frac{1}{V} dV \)
\(\textstyle W = -nRT \int\limits_{V_1}^{V_2} \frac{1}{V} dV \)
\(\textstyle W = -nRT (ln(V_2) – ln(V_1)) \)
\(\textstyle W = -nRT ln(\frac{V_2}{V_1}) \)
On peut rentrer le – dans le ln, ce qui donnerait :
\(\textstyle W = nRT ln(\frac{V_1}{V_2}) \)
Ces formules sont à redémontrer à chaque fois (mais comme tu le vois ce n’est pas trop compliqué).
On peut les résumer ainsi :
—
Pour une transformation :
isochore : W = 0
isobare : W = -PΔ V
isotherme : W = nRTln(V1/V2)
—
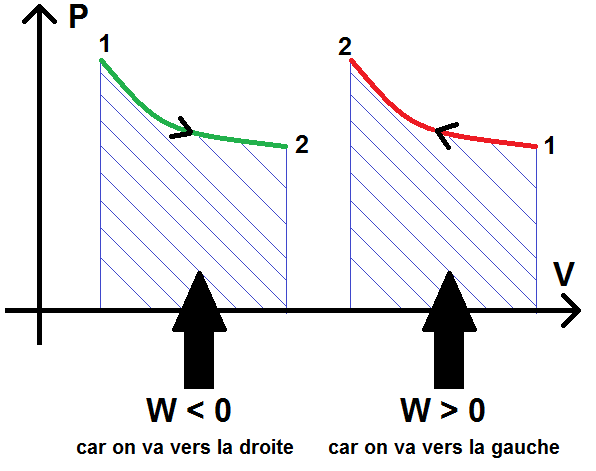
Ces résultats se retrouvent également grâce au diagramme de Clapeyron.
En effet, on a vu que ce diagramme comportait V en abscisse et P en ordonnée. Donc 
On peut en déduire W est l’opposé de l’aire sous la courbe de la transformation dans le diagramme de Clapeyron (opposé à cause du signe – dans le formule d W).
Concrètement, si on va de l’état initial à l’état final en allant vers la droite, W < 0, si on va vers la gauche W > 0.
En notant 1 l’état initial et 2 l’état final, cela donne :
Voyons à quoi correspondent les 3 transformations précédentes dans le diagramme (P , V) :
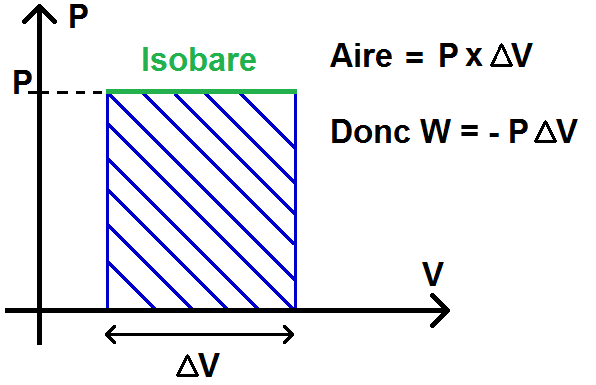
Remarque : ΔV peut éventuellement être négatif si on va de le droite vers la gauche
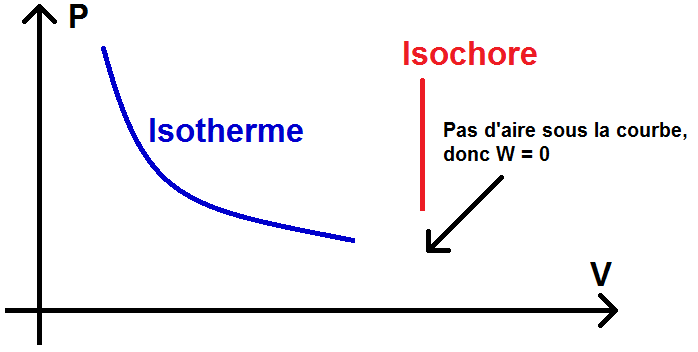
Pour la transformation isochore, la droite est verticale, donc l’aire sous la courbe est nulle : W = 0 : on retrouve le résultat précédent !
Pour la transformation isobare, la droite est horizontale, donc l’aire sous la courbe est un rectangle d’aire P(V2 – V1) = P ΔV, le travail étant l’opposé : W = -P ΔV : : on retrouve le résultat précédent !
Pour la transformation isotherme, la droite n’étant ni verticale ni horizontale, il n’y a pas de moyen de retrouver graphiquement la formule vue ci-dessus.
Nous allons maintenant pouvoir passer à la formule la plus importante du cours ! 
Le premier principe de la thermodynamique permet de relier W et Q que l’on a appris à calculer précédemment. Grâce au premier principe, nous allons pouvoir démontrer de nombreuses formules que nous détaillerons par la suite.
L’énoncé du premier principe de la thermodynamique est très simple :
\(\displaystyle \Delta U = W + Q \)
Dans cette formule :
ΔU est la variation d’énergie interne (en J)
W est la somme de tous les travaux (qui peuvent être négatifs)
Q est la somme de tous les transferts thermiques (qui peuvent être négatifs)
Ce principe signifie la conservation de l’énergie : celle-ci ne se perd pas mais se transforme en une autre forme (chaleur, travail…)
On peut évidemment écrire le premier principe sous forme infinitésimale :
\(\displaystyle dU = dW + dQ \)
Il suffira alors d’intégrer après avoir remplacé.
Attention petit rappel: quand on intègre dU cela donne ΔU car c’est une fonction d’état, mais quand on intègre dW et dQ cela donne W et Q car ce ne sont pas des fonctions d’état…
Un peu de vocabulaire supplémentaire maintenant : quand on dit qu’un système est isolé, cela signifie qu’il n’y a ni échange de matière ni échange d’énergie avec l’extérieur, donc W = 0 et Q = 0, donc d’après le 1er principe :
\(\displaystyle \Delta U = 0 \)
\(\displaystyle pour \, un \, système \, isolé \)
Nous verrons ci-dessous comment appliquer le premier principe, mais il faut tout d’abord introduire de nouvelles notions (et oui, encore du vocabulaire supplémentaire !!!  )
)
L’enthalpie, notée H, est exprimée en Joules. C’est une fonction d’état (on parlera donc de ΔH).
Ce n’est pas une nouvelle variable à proprement parler mais plus un regroupement de variables qui va nous permettre de simplifier les calculs et d’obtenir des propriétés intéressantes.
L’enthalpie est définie de la manière suivante :
\(\displaystyle H = U + PV \)
Comme tu le vois ce n’est pas vraiment une nouvelle variable puisqu’elle s’exprime en fonction de U, P et V.
Calculons dH :
\(\textstyle dH = dU + d(PV) \)
\(\textstyle dH = dU + PdV + VdP \)
Or d’après le 1er principe, dU = dQ + dW, et dW = -PdV
Donc dU = dQ – PdV. En remplaçant :
\(\textstyle dH = dQ – PdV + PdV + VdP \)
\(\textstyle dH = dQ + VdP \)
Voici donc l’expression infinitésimale de dH, que nous allons utiliser par la suite :
\(\displaystyle dH = dQ + VdP \)
Conséquence directe : pour une transformation isobare, dP = 0, donc dH = dQ, donc ΔH = Q.
Ainsi :
\(\displaystyle \Delta H = Q \)
\(\displaystyle pour\, une\, transformation\, isobare \)
Par ailleurs, les changements d’état se font à pression constante, donc ΔH = Q pour un changement d’état.
Mais on a vu précédemment que pour un changement d’état, Q = mL.
On a donc ΔH = mL, donc ΔH/m = L : lors d’un changement d’état, l’enthalpie massique (car on a divisé par m) correspond à la chaleur latente dont on a parlé précédemment, ce pourquoi cette chaleur latente est aussi appelée enthalpie de changement d’état.. A noter qu’elle peut être massique ou molaire.
Voyons maintenant un cas où l’on va utiliser dH.
On a vu précédemment que dQ = mcdT, avec c la capacité calorifique massique du corps considéré.
Elle correspond à la quantité d’énergie à apporter par échange thermique à un corps d’1 kg pour élever sa température de 1 degré (ou 1 Kelvin).
Par exemple, ceau = 4180 J.kg-1.K-1, donc il faut 4180 J pour augmenter la température d’1 kilogramme d’eau de 1 Kelvin.
Nous allons maintenant nous intéresser à la capacité calorifique C en J.K-1 (donc ni massique, ni molaire).
On avait vu comme précédemment que dQ = CdT.
Pour une transformation isochore, V = constante, on définit alors la capacité calorifique à volume constant, noté Cv, en J.K-1 de la manière suivante :
D’après le 1er principe, dU = dQ + dW.
Or V = constante, et on a vu dans ce cas que dW = 0.
Donc dU = dQ
Comme dQ = CvdT, on a dU = CvdT.
Ainsi :
\(\displaystyle C_v = \frac{dU}{dT})_v \)
Le )v signifie « à volume constant » (car transformation isochore).
De la même manière, pour une transformation isobare, P = constante, on définit alors la capacité calorifique à pression constante, noté Cp, en J.K-1 de la manière suivante :
d’après ce que l’on a vu sur l’enthalpie, dH = dQ + VdP.
Or P = constante, donc dP = 0.
Donc dH = dQ
Comme dQ = CpdT, on a dH = CpdT.
Ainsi :
\(\displaystyle C_p = \frac{dH}{dT})_p \)
Le )p signifie « à pression constante » (car transformation isobare).
On retrouve plus ou moins la même chose que pour Cv mais avec dH au lieu de dU.
Il y a donc 2 formules à retenir également que nous utiliserons dans de nombreux exercices :
\(\displaystyle dU = C_v dT \)
\(\displaystyle pour \, une \, transformation \, isochore \)
\(\displaystyle dH = C_p dT \)
\(\displaystyle pour \, une \, transformation \, isobare \)
A noter que même si la transformation n’est pas isobare ou isochore, ces relations restent vraies pour les gaz parfaits.
A partir de ces définitions, il existe une relation entre Cp et Cv pour les gaz parfaits.
En effet : H = U + PV
H = U + nRT car gaz parfait donc PV = nRT
dH = dU + d(nRT)
dH = dU + nRdT
Or dH = CpdT et dU = CvdT puisque l’on a un gaz parfait, d’où :
CpdT = CvdT + nRdT
En simplifiant par dT, on obtient ce que l’on appelle la relation de Mayer :
\(\displaystyle C_p = C_v + nR \)
On regroupe généralement les capacités calorifique ensemble :
\(\displaystyle C_p – C_v = nR \)
—
ATTENTION à ne pas inverser Cp et Cv dans cette formule !!
Pour retenir facilement, dis-toi que c’est dans l’ordre alphabétique, p est avant le v dans l’alphabet donc c’est Cp – Cv et non Cv – Cp.
—
nR étant positif, on en déduit que :
\(\displaystyle C_p \gt C_v \)
Nous allons voir maintenant qu’il existe d’autres formules avec Cv et Cp, que nous allons démontrer avec l’application principale de ces 2 coefficients : les transformations adiabatiques.
On considère un gaz parfait qui subit une transformation adiabatique, c’est-à-dire dQ = 0.
On a dU = dQ + dW = dW, avec dU = CvdT et dW = -PdV
Ce qui donne CvdT = -PdV
De même, dH = dQ + VdP = VdP
D’où CpdT = VdP
En divisant terme à terme les 2 équations, on a :
\(\textstyle \frac{C_p \, dT}{C_v \, dT} = \frac{VdP}{-PdV} \)
\(\textstyle \frac{C_p}{C_v} = \frac{VdP}{-PdV} \)
On regroupe alors les V à gauche et les P à droite pour obtenir une équation à variables séparées :
\(\textstyle -\frac{C_p}{C_v}\frac{dV}{V} = \frac{dP}{P} \)
Posons alors γ telle que :
\(\displaystyle \gamma = \frac{C_p}{C_v} \)
on a alors :
\(\textstyle -\gamma \frac{dV}{V} = \frac{dP}{P} \)
Il ne reste plus qu’à intégrer entre l’état initial et l’état final que nous noterons 1 et 2 (en sortant γ de l’intégrale qui est une constante) :
\(\textstyle -\gamma \int\limits_{V_1}^{V_2} \frac{dV}{V} = \int\limits_{P_1}^{P_2} \frac{dP}{P} \)
\(\textstyle -\gamma ln(\frac{V_2}{V_1}) = ln(\frac{P2}{P1}) \)
\(\textstyle \gamma ln(\frac{V_1}{V_2}) = ln(\frac{P2}{P1}) \)
\(\textstyle ln(\frac{V_1^\gamma}{V_2^\gamma}) = ln(\frac{P2}{P1}) \)
Il ne reste plus qu’à enlever les ln :
\(\textstyle \frac{V_1^\gamma}{V_2^\gamma} = \frac{P2}{P1} \)
En regroupant les variables de l’état 1 et de l’état 2 :
\(\displaystyle P_1V_1^\gamma = P_2V_2^\gamma \)
Que l’on peut également écrire sous forme :
\(\displaystyle PV^\gamma = constante \)
Cette formule constitue une des 3 formules de Laplace
Cette formule est très intéressante car elle permet de relier très simplement la pression et le volume entre l’état initial et l’état final.
La démonstration ci-dessus est à connaître mais tu n’auras pas à le refaire à chaque fois, tu peux directement utiliser la formule si tu sais que la transformation est adiabatique et que l’on a un gaz parfait.
Grâce à la loi des gaz parfaits, on peut très facilement trouver le même style de formule entre P et T, et entre T et V, qui constitueront les 2 autres formules de Laplace.
Faisons la démonstration pour T et V, tu pourras t’entraîner à la faire pour P et T
On repart de la formule démontrée ci-dessus :
\(\textstyle PV^\gamma = constante \)
Comme on veut la relation entre T et V, il faut faire disparaître P, donc le remplacer avec la loi des gaz parfaits, selon laquelle P = nRT/V, d’où :
\(\textstyle \frac{nRTV^\gamma}{V} = constante \)
\(\textstyle TV^{\gamma – 1} = \frac{constante}{nR} \)
nR étant constant, on peut remplacer constante/nR par une autre constante :
\(\displaystyle TV^{\gamma – 1} = constante \)
Et voilà, on vient de trouver la formule de Laplace entre T et V.
Pour celle entre P et T (que tu démontreras toi-même  ), on trouve :
), on trouve :
\(\displaystyle P^{1-\gamma}T^\gamma = constante \)
Comme tu le vois, celle-là est plus compliquée à retenir que les autres.
La plus simple étant PVγ = constante, c’est celle qui sera le plus utilisée dans les exercices, mais tu dois connaître les autres également, ainsi que la manière de les redémontrer !
Revenons aux coefficients Cv et Cp.
On a vu que :
\(\textstyle C_p – C_v = nR \)
\(\textstyle \frac{C_p}{C_v} = \gamma \)
On a 2 inconnues et 2 équations, on va pouvoir trouver l’expression de Cv et Cp !
On a Cp = γ Cv,
d’où γ Cv – Cv = nR
donc Cv(γ – 1) = nR
Ainsi :
\(\displaystyle C_v = \frac{nR}{\gamma – 1} \)
Et comme Cp = γ Cv :
\(\displaystyle C_p = \frac{nR\gamma}{\gamma – 1} \)
Remarque : : on a vu que Cp > Cv, donc γ > 1.
On a donc γ ≠ 1 (heureusement sinon on divise par 0 !), et γ – 1 > 0, donc tout est logique en terme de signe dans les formules ci-dessus.
—
ATTENTION à ne pas confondre les 2 formules qui se ressemblent énormément !!
En effet, seul le γ du numérateur différencie les 2 formules.
Pour retenir facilement, dis-toi que γ > 1, et Cp > Cv, donc le γ du numérateur est forcément pour le Cp.
—
Revenons sur les changements d’état.
Nous avons vu que tout corps pur peut exister sous 3 formes : solide, liquide, gaz.
L’état dans lequel se trouve le corps dépendra de sa pression et de sa température, on peut donc tracer les différentes zones d’existence dans un diagramme (P ; T) (à ne pas confondre avec le diagramme (P ; V)) :
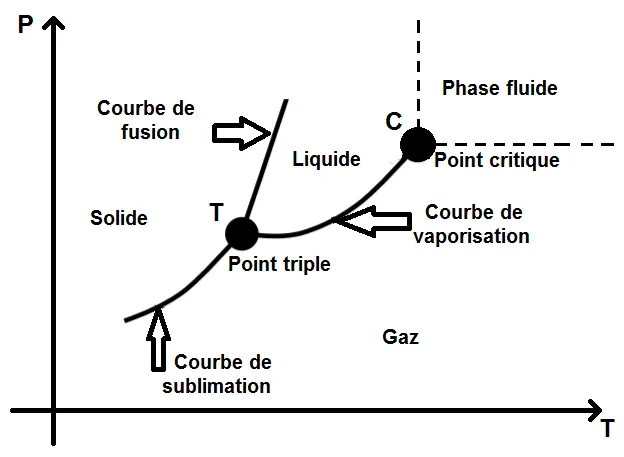
Dans ce diagramme, on voit les 3 zones d’existence du corps sous ses 3 états, séparés par 3 courbes : les courbes de fusion, de sublimation et de vaporisation correspondant aux changements d’état.
Ces 3 courbes se rejoignent au point triple, noté T : il s’agit de conditions de pression et de température sous lesquelles le fluide existe sous les 3 phases en même temps (c’est bizarre mais ça existe !).
Il y a également le point critique, noté C : au-delà de ce point, le corps entre dans une phase fluide, on parle de fluide super-critique : il n’est plus possible de faire la différence entre liquide et gaz (oui là aussi c’est très bizarre mais ça existe !).
La transition d’un état à un autre se fait à température constante mais aussi à pression constante, c’est-à-dire que rien ne varie dans le diagramme (P ; T) pendant le changement d’état, mais le volume change, ce pourquoi il est plus intéressant de regarder ce qui se passe dans le diagramme (P ; V) :
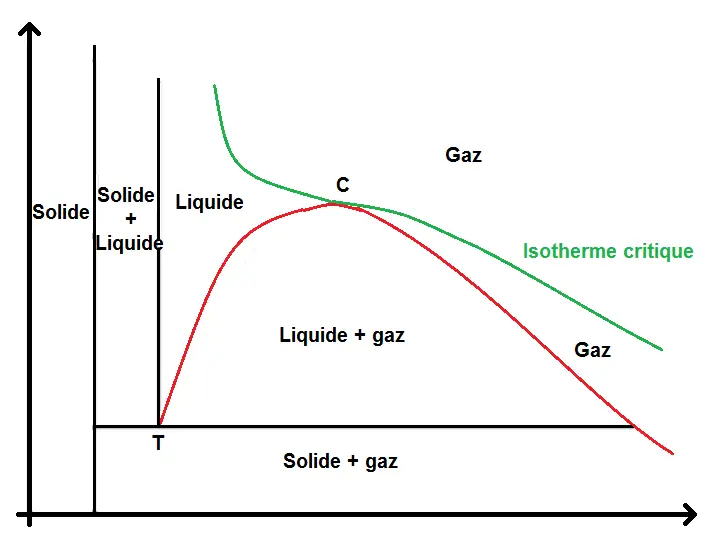
On retrouve le point triple T et le point critique C.
Les zones solide + gaz, solide + liquide et liquide + gaz sont les zones de changement d’état, dans lesquelles le corps va passer d’une phase à l’autre.
Nous allons nous intéresser particulièrement à l’équilibre liquide + gaz :
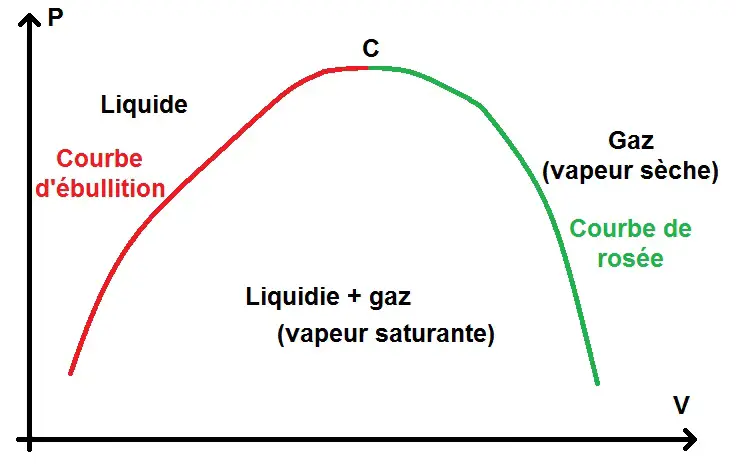
La courbe est décomposée en 2 parties : la courbe d’ébullition (aussi appelée courbe de bulle) et la courbe de rosée, séparées par le point critique C.
Quand il y a uniquement de la vapeur, on parle de vapeur sèche, quand elle est mélangée avec la phase liquide on parle de vapeur saturante ou vapeur humide (on parlera également de pression de vapeur saturante).
La zone d’équilibre liquide – gaz est appelée zone de saturation.
Il est intéressant de tracer les isothermes :
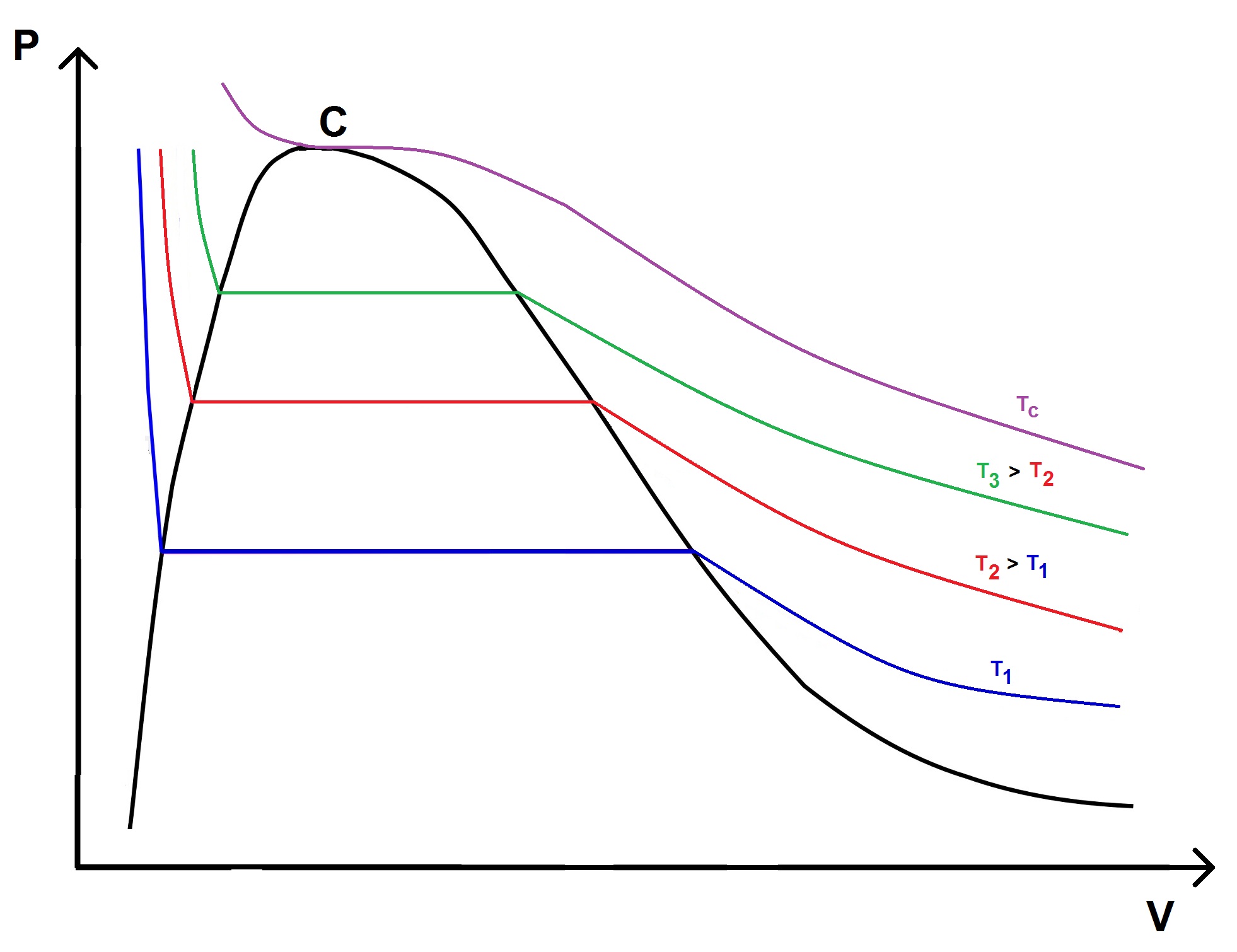
On voit que l’isotherme est horizontale lors du changement d’état quand la pression est constante.
—
Les changements d’état se faisant à pression constante se font également à température constante : cette pression est appelée pression de vapeur saturante.
—
Attention, quand le fluide se trouve dans la zone de saturation, il peut très bien changer d’isotherme (on verra cela en exercice).
Quand le fluide est dans la zone de saturation, on définit alors le titre en vapeur saturante : il s’agit de la proportion de masse de vapeur dans le mélange liquide + gaz (en gros le pourcentage de vapeur dans le mélange) :
\(\textstyle x = \frac{m_{vapeur}}{m_{vapeur} + m_{liquide}} \)
Mais ce qui va être utile pour les calculs, c’est le lien entre x et les volumes.
En effet, prenons un point dans la zone de saturation, et notons VL le volume du fluide quand il est sur la courbe d’ébullition, VV quand il est sur la courbe de rosée et V quand il est dans la zone de saturation :
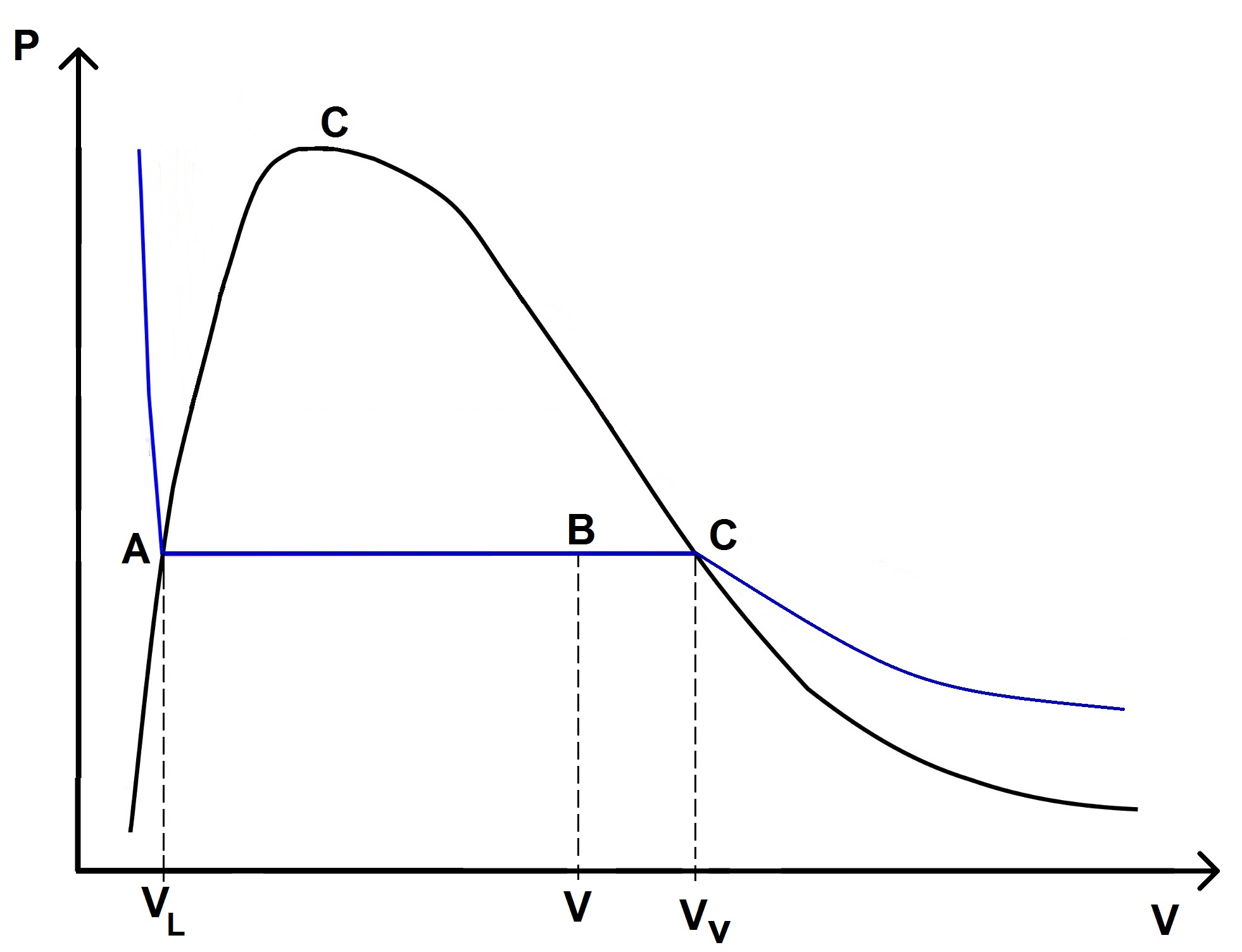
x étant le pourcentage de vapeur, 1 – x est le pourcentage de liquide, donc on a :
\(\textstyle V = xV_v + (1 – x)V_L \)
\(\textstyle V = xV_v + V_L – xV_L \)
\(\textstyle V – V_L = x(V_v – V_L) \)
\(\displaystyle x = \frac{V – V_L}{V_v – V_L} \)
Or sur le graphique, V – VL correspond à la distance AB, tandis que VL – VL correspond à la distance AC.
\(\displaystyle x = \frac{AB}{AC} \)
Remarque : la formule ci-dessus est également valable pour l’énergie interne,l’enthalpie et l’entropie (notée S et que l’on verra dans le chapitre sur le second principe) :
\(\textstyle x = \frac{V – V_L}{V_v – V_L} = \frac{U – U_L}{U_v – U_L} = \frac{H – H_L}{H_v – H_L} = \frac{S – S_L}{S_v – S_L} \)
Autre remarque : x = 0 correspond au fluide entièrement sous forme liquide (sur la courbe d’ébullition), et x = 1 au fluide entièrement sous forme gazeuse (sur la courbe de rosée).
Avant de terminer ce chapitre, voyons une dernière petite chose très simple.
Dans les exercices, il y aura parfois à traduire l’équilibre mécanique d’une paroi ou d’un piston par exemple, en disant que les forces se compensent.
Dans ce chapitre, il faut prendre en compte les forces dues à la pression des gaz (aussi appelées forces pressantes), qui se calculent très simplement par la formule :
\(\displaystyle F = P \times S \)
Cette formule n’est valable que si la pression est constante sur toute la surface de contact, ce qui sera presque toujours le cas, sinon il faut utiliser la formule dF = PdS et intégrer.
Dans cette formule, F est la force pressante en N, la pression du gaz en Pa, S la surface de contact en m2.
Attention, il faudra souvent convertir la surface ainsi que la pression, fréquemment donnée en bars et non en Pa.
Si on veut mettre F sous forme de vecteur, on rajoute un vecteur unitaire perpendiculaire à la surface de contact.
La force s’exerce sur toute la paroi mais on regroupe tout en une seule force qui s’applique au centre de la paroi.
Exemple :
On a une paroi cylindrique de surface S qui coulisse dans un axe. Un gaz est situé dans la partie gauche, à la pression P1, l’atmosphère extérieure est à la pression P0 :
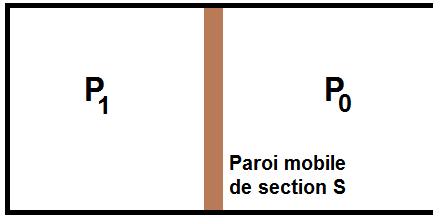
Le gaz à l’intérieur exerce une force horizontale dirigée vers la droite, de norme F1 = P1S.
L’air extérieur exerce une force horizontale dirigée vers la gauche, de norme F0 = P0S :
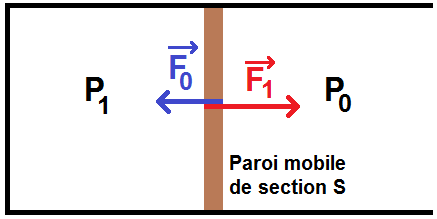
Ainsi, s’il n’y a pas d’autre force, si P1 > P0, la paroi se déplacera vers la droite, sinon elle ira vers la gauche.
Sur l’exemple ci-dessus, P1 > P0 donc F1 > F0 : la paroi ira vers la droite.
Ce premier chapitre sur la thermodynamique est désormais terminé  mais il y a d’autres chapitres sur la thermo !
mais il y a d’autres chapitres sur la thermo !
Il y a eu dans ce cours beaucoup de nouvelles formules et de nouveau vocabulaire, ce pourquoi il faut faire beaucoup d’exercices, en commençant par des exercices basiques d’application directe du cours et en complexifiant au fur et à mesure.
Tu as de la chance, il y a justement une page avec de nombreuses applications basiques des propriétés et formules vues précédemment, il est disponible en cliquant ici.
Une fois tout cela bien maîtrisé, tu pourras passer au 2ème principe de la thermodynamique (rassures-toi, il n’y en a que 2 !) puis à l’étude des machines thermiques.
Des exercices en vidéo disponibles ci-dessous t’aideront également à bien comprendre le chapitre
Les exercices sur ce chapitre seront bientôt disponibles !
En attendant, tu trouveras ici des exercices basiques sur la thermodynamique.
Sommaire des coursHaut de la page


Salut, merci pour le cours, mais peut-on revoir la partie sur le signe du travail (W) en fonction du diagramme de Clapeyron?
Salut, merci pour le cours, mais peut on revoir la partie sur le signe du travail en fonction du diagramme de Clapeyron?
Bonjour, c’est un super cours très accessible. Même à un novice comme moi en prenant mon temps. Cependant j’écris surtout ce message car j’ai une petite interrogation :
Dans la partie calcul de W, sous partie transformations isotherme, lors le calcul de l’intégral. Quand on sort – nRT de l’intégrale, il y a toujours un signe moins à l’intérieur. Ça me parait illogique, de plus il disparait à la ligne du dessous, est-ce une erreur du cours ou de ma part.
Merci d’avance
Bonjour merci beaucoup, vous me sauvez. Votre cours est très bien fait et simple à comprendre.
Merci infiniment pour ce cours,
Très clair et simple
J ai bien compris la thermo grace à vous
Merci bcp
Merci, cours très complet et intéressant !
Mais tout cela est-il exigible au programme officiel ou y a t il des suppléments ?
Merci ! Normalement tout est exigible mais tout dépend de ton école/ cursus.